Was ist eine kryoglobulinämische Vaskulitis? Was ist Schönlein-Genoch-Purpura? Was ist Wegener-Granulomatose? Was ist eine granulomatöse Angiitis nach Cherdzha-Strauss? Was ist eine mikroskopische Polyangiitis? Die Antworten finden Sie in diesem Artikel..
Inhalt
Kryoglobulinämische Vaskulitis
Die essentielle kryoglobulinämische Vaskulitis ist eine Erkrankung, bei der kleine Gefäße durch kryoglobulinämische Komplexe mit Schädigung der Haut und der Nierenglomeruli geschädigt werden. Kryoglobuline sind Immunglobuline oder immunoglobulinhaltige Komplexe, die bei niedrigen Temperaturen spontan ausfallen und ein Gel bilden; wenn die temperatur steigt, werden sie wieder löslich.
Die Ursache der kryoglobulinämischen Vaskulitis kann eine verlängerte Hypothermie, Hepatitis B- und C-Viren sein.
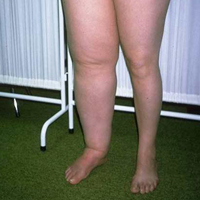
Vor allem Frauen über 50 Jahre leiden an einer kryoglobulinämischen Vaskulitis. Leitsymptom ist ein Hautausschlag (tastbare Purpura) auf der Haut der Beine und Füße, begleitet von Juckreiz oder Brennen. Hautveränderungen können durch Kälteeinwirkung, Druck auf die Haut oder längeres Stehen verursacht werden. Das Auftreten eines Hautausschlags wird von Schmerzen in den Muskeln und Gelenken begleitet. Anstelle von Hautpurpura können sich Nekrose und trophische Geschwüre (insbesondere an den Beinen) entwickeln. Mögliche Nierenschäden wie Glomerulonephritis und Lungen mit Kurzatmigkeit und Hämoptyse.
Die Diagnose wird durch den Nachweis von Kryoglobulinen im Blutserum bestätigt.
Purpura Schönlein-Genoch (hämorrhagische Vaskulitis)
 Die hämorrhagische Vaskulitis (Schönlein-Henoch-Krankheit) ist eine systemische Gefäßerkrankung mit einer vorherrschenden Läsion von Kapillaren, Arteriolen und Venolen, hauptsächlich der Haut, Gelenke, Bauchhöhle und Nieren.
Die hämorrhagische Vaskulitis (Schönlein-Henoch-Krankheit) ist eine systemische Gefäßerkrankung mit einer vorherrschenden Läsion von Kapillaren, Arteriolen und Venolen, hauptsächlich der Haut, Gelenke, Bauchhöhle und Nieren.
Die Erkrankung tritt meist bei Kindern und Jugendlichen, seltener bei Erwachsenen beiderlei Geschlechts, nach einer Infektion (Streptokokken-Halsschmerzen oder Verschlimmerung einer Mandelentzündung, Pharyngitis), der Verabreichung von Impfstoffen und Seren sowie im Zusammenhang mit Medikamentenunverträglichkeit, Kühlung auf.
Die wahrscheinlichste autoimmune Natur der Krankheit. Jene. Gefäßzellen beginnen als körperfremder Wirkstoff aufzusteigen und werden von körpereigenen Schutzzellen angegriffen.
Die Krankheit manifestiert sich durch eine Trias von Symptomen: hämorrhagische Hautausschläge (Purpura), Gelenkschmerzen (Arthralgie) und (oder) Gelenkentzündungen (Arthritis), hauptsächlich große Gelenke, und ein Bauchsyndrom, das bei 70% der Patienten auftritt. Die letzten beiden Anzeichen werden normalerweise von Fieber begleitet. Die häufigste kutane (einfache) Variante ist durch Hautausschläge mit symmetrischen Ausschlägen an den Gliedmaßen, am Gesäß und seltener am Rumpf gekennzeichnet. Beim Drücken verschwinden die Elemente des Hautausschlags nicht. Die artikuläre Variante tritt zusammen mit der kutanen einen oder wenige Tage danach auf und äußert sich durch Schmerzen in großen Gelenken. Nach ein paar Tagen verschwinden die Schmerzen, können aber bei einer neuen Welle wieder auftreten. Das Abdominalsyndrom ist durch hämorrhagische Eruptionen in der Schleimhaut des Gastrointestinaltrakts, Mesenterium, Peritoneum gekennzeichnet, die in einigen Fällen zu Ulzerationen und Perforationen mit der Entwicklung einer Peritonitis führen. Klinisch manifestiert es sich durch plötzliche Schmerzen der Art von Darmkolik, Erbrechen und Durchfall mit Blut, die einer abdominalen Pathologie anderer Art ähneln (Peptidgeschwür, Colitis ulcerosa, Ruhr). Das Abdominalsyndrom heilt normalerweise in 2-3 Tagen ab. Oft gibt es eine Kombination der abdominalen Form mit Nierenschäden, die sich durch das Auftreten von Protein und Blut im Urin manifestieren. Am gefährlichsten ist die Blitzform. Die extreme Schwere der fulminanten Form ist auf die generalisierte Natur des Hautausschlags zurückzuführen, der oft zum Tod durch Blutungen im Gehirn und seinen Membranen führt.
Die Diagnose basiert auf dem Vorhandensein einer charakteristischen Trias von Anzeichen oder nur hämorrhagischen Hautausschlägen.
Wegener-Granulomatose
Die Wegener-Granulomatose ist eine entzündliche Gefäßerkrankung mit einer vorherrschenden Läsion von Arterien und Venen mittleren und kleinen Kalibers sowie Gefäßen der Mikrogefäße der Atemwege, Lunge und Nieren..
Unter den möglichen Ursachen der Wegener-GrV-Granulomatose wird ein mikrobieller oder viraler Faktor als wahrscheinlicher angesehen. Die Krankheit tritt häufig nach ARVI, Hypothermie, Impfung, Antibiotikatherapie auf, kann sich aber auch bei gesunden Kindern entwickeln.
Die Krankheit beginnt in jedem Alter, aber häufiger in 40-45 Jahren. Die oberen Atemwege sind betroffen - Ulzerationen, Nekrose treten auf; lunge - infiltriert mit Verfall; Nieren - Glomerulonephritis. Es gibt zwei Formen der Wegener-Granulomatose - lokal und generalisiert.
Die ersten Beschwerden bei der Wegener-Granulomatose sind Kopfschmerzen, Ohren- und Mundschmerzen, Nasenbluten, Hämoptyse, Schwellung der Augenlider und laufende Nase. Mit der Niederlage der HNO-Organe, einer anhaltenden Rhinitis mit blutig-eitrigem Ausfluss, tritt ein Bild einer Sinusitis auf, die Schleimhaut der Nasengänge, der weiche Gaumen, die Mandeln sind nekrotisch. Dann kommt es zu einer Zerstörung der Nasenscheidewand, Concha mit Perforation des weichen und harten Gaumens. Röntgen der Lunge zeigt beidseitig abgerundete Infiltrate mit Karies und Kariesbildung.
Bei der generalisierten Form der Wegener-Granulomatose tritt nach einigen Wochen oder Monaten vor dem Hintergrund eines fortschreitenden nekrotischen Prozesses ein Hautsyndrom im Vordergrund - verschiedene, knotige, hämorrhagische und ulzerative nekrotische Elemente, einige Patienten entwickeln eine Myokarditis, Perikarditis, Bronchitis.
Allerdings ist die Glomerulonephritis, die in diesem Stadium eintritt, prognostisch am beeindruckendsten. Es verläuft ohne arterielle Hypertonie mit der schnellen Entwicklung eines Nierenversagens..
Granulomatöse Angiitis von Cherd-Strauss
Allergische Vaskulitis oder granulomatöse Angiitis nach Churg-Strauss ist eine entzündliche Erkrankung kleiner und mittlerer Gefäße, hauptsächlich der Haut, der peripheren Nerven und der Lunge. Menschen im Alter von 30-40 Jahren werden krank.
Die Krankheit ist durch ein klinisches Bild von Asthma bronchiale und eine Zunahme der Anzahl von Eosinophilen im Blut gekennzeichnet. In der Anfangsphase haben die Patienten verschiedene allergische Manifestationen, darunter eine laufende Nase, Heuschnupfen und Asthma bronchiale..
Das Churg-Strauss-Syndrom wird empfohlen, wenn eine Person mit einer Vorgeschichte von Allergien oder Asthma bronchiale eine Erhöhung der Anzahl von Eosinophilen im Blut in Kombination mit einer systemischen Erkrankung aufweist, bei der Infiltrate in der Lunge (70%) oder Schäden an mehrere Organe. Die Diagnose wird im Krankenhaus durch die Ergebnisse einer Biopsie von geschädigtem Gewebe bestätigt.
Mikroskopische Polyangiitis
Die mikroskopische Polyangiitis (Polyarteriitis) ist eine entzündliche Gefäßerkrankung, die die Kapillaren, Venolen und Arteriolen der Lunge, der Nieren und der Haut befällt.
Die mikroskopische Polyangiitis wird hauptsächlich bei Männern mittleren Alters beobachtet. Das Auftreten von Fieber, das gegen eine Antibiotikatherapie resistent ist, Schwäche, Gewichtsverlust, Gelenkschmerzen wird festgestellt. Die Haut kann einen Ausschlag mit ulzerativen Veränderungen haben. Lungenschäden entwickeln sich mit Kurzatmigkeit, Hämoptyse und möglicherweise schweren Lungenblutungen. Im Gegensatz zur Wegener-Granulomatose findet man in der Lunge röntgenologisch Infiltrate ohne Zerfall. Die Entwicklung einer hämorrhagischen oder schnell fortschreitenden fibrosierenden Alveolitis ist möglich. Die Niederlage der Nierenglomeruli äußert sich durch das Auftreten von Protein- und Blutverunreinigungen im Urin, die Entwicklung eines nephrotischen Syndroms ist möglich. Bei schnell fortschreitender Glomerulonephritis entwickelt sich nach 3-6 Monaten ein Bild eines Nierenversagens.