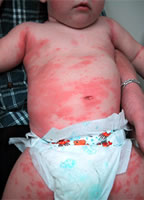
Die primäre Immunschwäche ist eine angeborene Störung des Immunsystems, die sich als Folge genetischer Störungen entwickelt. Die Formen der primären Immunschwäche sind ein Mangel des Komplementsystems, Phagozytose sowie humorale und zelluläre Immunität..
Inhalt
Das Konzept der primären Immunschwäche
Primäre Immundefekte sind angeborene Erkrankungen des Immunsystems, die mit genetischen Defekten in einer oder mehreren Komponenten des Immunsystems verbunden sind, nämlich Komplement, Phagozytose, humorale und zelluläre Immunität. Allen Formen der primären Immunschwäche gemeinsam ist das Vorliegen chronischer Infektionen, die verschiedene Organe und Gewebe betreffen und in der Regel durch opportunistische Mikroflora verursacht werden. Opportunistische Infektionen sind eine große Gruppe verschiedener Infektionskrankheiten. Was sie eint, ist, dass sie nur mit einer ausgeprägten Abnahme der Immunität auftreten. Bei einer gesunden Person oder einer Person mit einem guten Immunitätszustand treten diese Krankheiten in der Regel nicht auf oder sind mild..
Nach dieser Klassifikation werden primäre Immundefekte in 5 Gruppen eingeteilt:
- Mangel an humoraler Immunität
- Insuffizienz der zellulären Immunität
- kombinierter Mangel an humoraler und zellulärer Immunität
- Phagozyteninsuffizienz
- Komplementmangel
Die moderne Klassifikation der primären Immundefekte basiert auf der vorherrschenden Niederlage der einen oder anderen Immunität..
Mangel an Phagozytose
Die Insuffizienz der Phagozyten beträgt 10-15% aller primären Immundefekte. Die Insuffizienz von Phagozyten wird durch eine gestörte Proliferation (Proliferation), Differenzierung, Chemotaxis (motorische Reaktion von Zellen als Reaktion auf die Einwirkung von Chemikalien) von Neutrophilen und Makrophagen verursacht - Zellen, die aktiv an der Phagozytose beteiligt sind, und tatsächlich eine Verletzung des Phagozytoseprozesses.
Phagozytäre Zellen (an der Phagozytose beteiligte Zellen) werden durch Leukozyten repräsentiert, und Makrophagenzellen spielen eine wichtige Rolle beim Schutz des Körpers vor pyogenen Bakterien und anderen intrazellulären Mikroorganismen. Ein schwerer Mangel an polymorphkernigen Leukozyten (Neutropenie - eine Abnahme der Anzahl von Neutrophilen im allgemeinen Bluttest) kann zur Entwicklung einer massiven bakteriellen Infektion führen.
Von besonderer Bedeutung sind zwei genetische Defekte, die die Funktion von Fresszellen stören, die mit dem Auftreten schwerer Krankheiten verbunden sind, oft mit tödlichem Ausgang - chronische Granulomatose (deren Ursache eine Verletzung des Sauerstoffrückgewinnungsmechanismus ist) und unzureichende Adhäsion ( Adhäsion) von Leukozyten (aufgrund von Gendefekten).
Insuffizienz des Komplementsystems
 Die Insuffizienz des Komplementsystems beträgt nicht mehr als 2% aller primären Immundefekte, äußert sich in einer Verletzung der Opsonisierung (dem Prozess der Interaktion mit Bakterien), der Phagozytose (dem Prozess des Einfangens und Verdauens von Mikroorganismen) und der Zerstörung von Mikroorganismen und wird begleitet durch schwere Infektionen bis hin zur Entwicklung einer Sepsis. Komplementdefekte treten häufig bei Autoimmunerkrankungen wie systemischem Lupus erythematodes auf.
Die Insuffizienz des Komplementsystems beträgt nicht mehr als 2% aller primären Immundefekte, äußert sich in einer Verletzung der Opsonisierung (dem Prozess der Interaktion mit Bakterien), der Phagozytose (dem Prozess des Einfangens und Verdauens von Mikroorganismen) und der Zerstörung von Mikroorganismen und wird begleitet durch schwere Infektionen bis hin zur Entwicklung einer Sepsis. Komplementdefekte treten häufig bei Autoimmunerkrankungen wie systemischem Lupus erythematodes auf.
Der Mangel an Komponenten des klassischen Weges der Komplementaktivierung bewirkt eine Prädisposition für Erkrankungen, die durch Störungen der Bildung von Immunkomplexen verursacht werden, beispielsweise für das Auftreten von systemischem Lupus erythematodes. Ein Komplementmangel führt zu einer erhöhten Empfindlichkeit des Körpers gegenüber eitrigen Infektionen. Eine Insuffizienz der terminalen Komponenten des Komplementsystems sowie der Komponenten des alternativen Weges schafft eine besondere Prädisposition für Infektionen - Gonorrhoe und Meningitis. Dies zeigt deutlich die wichtige Rolle des alternativen Weges der Komplementaktivierung und der destruktiven Membran des Komplexes bei der Entfernung von Bakterien dieser Spezies..
Die schwersten Verletzungen der Komplementfunktionen sind mit einem Mangel an Komponenten des Komplementsystems verbunden, der zu einem erblichen Angioödem führt (eine allergische Reaktion, die von Ödemen der Mundschleimhaut und der oberen Atemwege begleitet wird); diese Krankheit wird autosomal-dominant vererbt. Ein Inhibitor des Komplementsystems blockiert nicht nur den klassischen Weg der Komplementaktivierung, sondern hemmt auch die Aktivität der damit verbundenen Elemente des Blutgerinnungssystems. Grundsätzlich werden Formen des Mangels an Komplementkomponenten autosomal-rezessiv vererbt..